Top down proteomics is still tough -- some of the problems that were really hard 10 years ago are still really hard now. What if we could simplify the whole thing by doing something completely different?
Like this...?!?!?!?

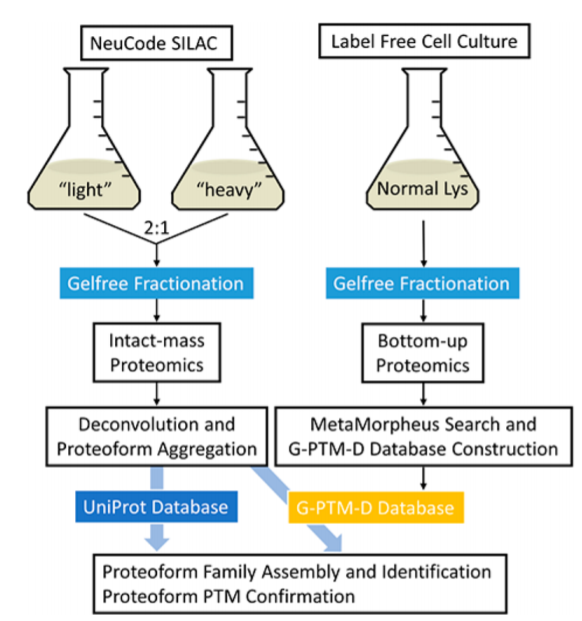
What if you looked at the challenge of trying to work out an intact protein sequence (including PTMs!) from the MS/MS spectra and set that to the side for now? Instead you use a combination of the intact protein masses alone (those are much easier to get!) and you combine that with ultra-deep shotgun analysis to work out the PTMs? Could you then link the proteoforms back together?
Some of them for sure, but there is going to be a whole lot of uncertainty there between proteoforms of similar mass. What if you knew something really cool about the intact proteins that would help you link it back to the shotgun measurements -- like EXACTLY how many lysines are in each protein!
This is yet another thing that you can do with the NeuCode reagents. The application of NeuCode in this manner was previously shown in this paper by many of these authors.

Honestly, it sounds like a neat trick -- TADAA! this is how many lysines are in this protein, but there are other ways we could do this, right?
What this new study does is shows how we can actually apply this to a biological system -- by delivering the largest number of E.coli PTM-annotated proteoforms we've ever seen in a single analysis (>500). It is worth noting that there are some of the familiar top-down limitations, like proteins >45kDa were excluded from analysis, but what a cool new method to have in our utility belts!